遗传病论文(独家整理6篇),索要一篇关于人类遗传疾病的论文
遗传病论文(独家整理6篇)
寻找人类遗传病的论文遗传病是指由遗传物质或致病基因控制的疾病,通常具有垂直传播和终身传播的特点。因此,遗传病具有从父母传给后代的特点。这种传播不仅指疾病的传播,也指致病基因在最基本层面的传播。因此,遗传病的发生表现出一定的家族特征。父母的生殖细胞。

求关于遗传病的论文 2500字
疾病简介遗传病是指由遗传物质变化引起的疾病或由致病基因控制的疾病。由遗传物质变化引起的疾病,包括染色体畸变和染色体水平上看不见的基因突变,统称为遗传病。根据所涉及的遗传物质的变化过程,遗传病可分为遗传病。你可以参考其他人在《亚洲遗传病案例研究》杂志上发表的论文,你必须拥有一切。检查人群中的遗传疾病。东莞中学任彩棉523005的教学目标:1。知识目标(1)通过调查分析,使学生了解一些人类遗传疾病的发病率;(2)通过对调查结果的分析,学生可以加深对遗传病的理解和对遗传病危害的认识。 2.能力目标(1)通过调节,遗传病研究和诊断性脱氧核糖核酸分析是重要的发展手段,其中限制性片段长度多态性(RFLP)分析在遗传病判断中应用最为广泛 遗传病的临床诊断比其他疾病更困难。 一方面,有许多种遗传病;另一方面,每种遗传病的个体发病率很低,因此遗传病的临床诊断是不可接受的。我也是一名湖泊医生。我把两本书搬出了图书馆,并学了它们。我和你的论文有相同的主题
索要一篇关于人类遗传疾病的论文
寻找人类遗传病的论文遗传病是指由遗传物质或致病基因控制的疾病,通常具有垂直传播和终身传播的特点。因此,遗传病具有从父母传给后代的特点。这种传播不仅指疾病的传播,也指致病基因在最基本层面的传播。因此,遗传病的发生表现出一定的家族特征。父母的生殖细胞。
求关于遗传病的论文 2500字
遗传病论文(独家整理6篇)范文
关于遗传疾病的第一篇论文(1)
标题:LRP4和罕见人类遗传疾病
摘要:2010年,低密度脂蛋白受体相关蛋白4(低密度脂蛋白受体相关蛋白4,lrp4)被报道并确认为cls-并指综合征(Cenani-Lenz syndactyly syndrome)的致病基因。文献报道,LRP4的突变可导致17型先天性肌无力综合征(迈锡尼综合征,认知17型)和2型硬化症(硬化症2型)。LRP4参与经典WNT信号通路和MAPK/JNK信号通路的调节,并与MUSK/AGRIN在神经肌肉接头处形成复合体来调节突触后转化。LRP4参与肢体末端、神经肌肉接头、肾脏、乳房和牙齿的发育和形成,并参与骨代谢过程。本文将重点介绍人类由LRP4引起的三种单基因疾病,并从基因发育的角度综述LRP4的研究进展,为进一步研究LRP4在脊椎动物早期发育中的作用机制提供参考。
关键词:LRP4CLS并指;先天性肌无力;ⅱ型硬化性骨病;信号路径;

LRP4与人类罕见遗传病
邵王金会刘志豪丛柳兆田杰京
西北大学教育部西部资源生物学与生物技术重点实验室
摘要:
4基因(LRP4)被发现与低密度致病脂蛋白受体相关。最近的蛋白质也被鉴定为Cenani-Lenz syndactyly myathenic综合征JNK基因,2010年。研究涉及LRP4的突变导致先天性信号17和硬化症2。LRP4在调节规范结合WNT途径和MAPK/信号途径;在神经肌肉连接处,LRP4与MUSK/AGRIN连接处,复杂的乳腺调节腺体突触后牙齿的转化。摘要:骨转移蛋白4参与四肢、过程、神经肌肉肾和发育,以及骨转移蛋白4和新陈代谢。本文总结了三种人类将提供罕见的遗传参考疾病的最新发展机制和早期骨转移蛋白4的遗传学研究。
关键词:
LRP4CLS综合症;先天性肌肉感觉综合征;硬化症2;信号通路;
1低密度脂蛋白受体家族和LRP4蛋白结构
低密度脂蛋白受体家族是一大组进化上保守的跨膜蛋白,其成员可以在包括蛔虫、果蝇和脊椎动物在内的各种物种中找到[1]。哺乳动物中有9种低密度脂蛋白受体,包括LRP1、LRP1B、巨蛋白(LRP29)、LDLR、VLDLR、LRP4、LRP5、LRP6和Apo ER2 (LRP8),它们具有共同的结构域,包括胞外表皮生长因子(人表皮生长因子)前体重复序列、跨膜区和胞质尾区[2]。LDLR首先被认为在受体介导的内吞过程中将脂蛋白作为内吞受体转运到细胞中。在这个过程中,特殊的配体和相应的受体结合在细胞表面,进入细胞,然后释放到细胞的其他部分。低密度脂蛋白受体主要调节细胞外基质中脂蛋白的浓度,并将它们转运到细胞中。大量研究表明,低密度脂蛋白受体家族成员也可以作为许多信号转导途径中的直接传导或调节因子,如LRP5(低密度脂蛋白受体相关蛋白5)和LRP6(低密度脂蛋白受体相关蛋白6)可以作为Wnt信号转导途径[3,4]的共受体。
低密度脂蛋白受体相关蛋白4是低密度脂蛋白受体家族的重要成员。中山等人[5]在哺乳动物细胞中发现了一种具有多个表皮生长因子样基序的跨膜蛋白。人LRP4基因位于11号染色体短臂1区1带2子带(11p11.2)中,含有1905个氨基酸,其中约90%是细胞外区,只有8%是细胞内区。它的胞外区(ECD)由一个信号肽、8个LDLα区(a型重复区)、4个β折叠区(b型重复区,或YWTD区)和1个O-连接寡糖修饰区组成。紧邻跨膜区的胞内区(ICD)包含Npx Y(蛋白质基序)结构单元和PDZ(盘状同源区)相互作用单元。此外,LRP4有6个表皮生长因子结构域,其中2个位于最后一个LDLα区和第一个β折叠区之间,3个位于每个β折叠区之间,1个位于跨膜区[2]之前。LRP4的结构与LRP5和LRP6非常相似,但它们的功能不同。LRP5/LRP6是经典Wnt信号通路的次级受体,其促进通路的激活,而LRP4被认为拮抗经典Wnt信号通路并抑制通路[6]的激活。
2 LRP4和罕见人类遗传疾病
目前,对LRP4的研究主要集中在骨骼和神经上。LRP4基因的不同突变可导致不同的遗传性疾病,主要包括CLS并指综合征(Cenani-Lenz syndactyly syndrome);CLSS,MIM 212780 [7],硬化症2;SOS,MIM 614305 [8]和17型先天性肌无力综合征(肌强直综合征,先天性17;[9].
2.1 LRP4和CLS并指
CLS并指症是一种常染色体隐性疾病,由两位医生塞纳尼·阿和伦茨·威于1967年在[首次报道。他们报道了一对兄弟,他们的情况类似于阿普特综合征[常染色体隐性遗传,特征是手、足和趾(趾),颅缝早期闭合,面中部发育不良],但不同的是,除了趾外,患者还患有桡尺缩短、融合、掌骨融合、指(趾)骨发育障碍、足受累但症状轻微,是一种新型的趾(趾)综合征。随后,他们将兄弟俩的症状分为利伯曼(1938)[(11)、博尔斯基(1958)[(12)和伊尔顿(1962)[(13)报告的一类并指定了他们的症状,并将其改名为塞纳-伦茨(趾)综合征(CLSS) [10]。CLS并指非常罕见,到目前为止只发现了22个家族,[7,14,15,16,17]。CLS并指患者的典型特征是:手指(趾)骨与四肢掌骨的缩短融合,少趾趾骨周围皮肤组织的融合。这种肢端肥大症不仅表现在手指(趾)骨和手掌(跖骨)骨,还影响手腕(跗骨)骨和上肢及下肢远端。如腕(跗)骨脱位、尺骨和桡骨缩短和融合,甚至脊柱发育不良、身材矮小等。,上肢的症状比下肢的症状更严重。一些病人也有轻微的面部异常,如上脸下垂、前额突出、额骨发育不良和眼睛距离宽。通过对已报道的CLS患者的表型分析,在大多数患者中也发现肾发育不全或功能障碍[18]。
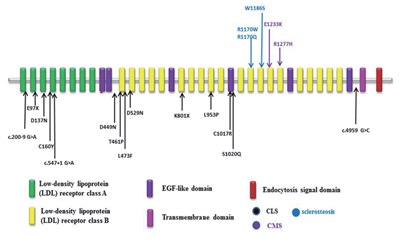
在2010年,李等人[7]首次确定人类LRP4的功能缺陷是CLS并指的主要原因。通过对14个CLS家系主要家系成员所有外显子的测序和基因筛选,最终确定CLS并指致病基因为LRP4。本文总结了CLS并指的不同亚型,大多数患者伴有肾发育不良,通过双荧光素酶实验分析,导致CLS并指的Lrp4(小鼠LRP4同源基因)错义点突变蛋白不能拮抗经典Wnt信号通路。野生型Lrp4蛋白可拮抗Wnt1(小鼠Wnt家族成员1)和LRP6(小鼠Lrp6同源基因)(均使用小鼠基因)激活的经典Wnt信号通路,还发现导致CLS并指的Lrp4错义点突变蛋白不能正常转运至细胞膜[7]。在缺乏Lrp4的小鼠中,发现在不同类型的缺乏Lrp4的小鼠中,包括Lrp4ECD/ECD、Lrp4mte/mte、Lrp4mitt/mitt、Lrp4mitt/mdig和Lrp4mdig/mdig,都伴随有并指,并且一些甚至在整个四肢如腕骨中具有发育缺陷。据报道引起CLS并指的LRP4突变位点和突变类型在图1中用黑色箭头表示。
大多数CLS并指患者伴有肾发育不良[7],这表明LRP4也参与肾发育。同时,在Lrp4缺陷小鼠[20,22,23,24中也发现伴随肾发育不良的表型。Tanahashi等人图1 LRP4的结构图和LRP 4报道的突变位点构建了Lrp4-/-敲除小鼠,这些小鼠在胚胎期死亡。敲除小鼠胚胎的检测显示敲除小鼠具有双侧肾脏缺陷,并且胚胎具有羊水过多和羊水清除途径缺陷。
2.2 LRP4和2型硬化性骨病
硬化症2也是一种常染色体隐性疾病,是一种由骨过度生长引起的严重硬化性骨发育疾病。布埃诺等人于1994年首次提出这一建议。他们发现一名17岁的西班牙患者在12岁时患有右侧面神经麻痹。检查发现瘫痪是由面部不对称引起的,前额有轻微硬块,脸中部发育不良,牙齿咬合不正,前额突出。出生后双侧3~4个手指和脚趾有并指(趾);此外,远端手指缩短并径向偏移,指甲发育不良。影像学检查结果显示颅骨大面积均匀硬化,静脉明显,鞍点和额窦增大,下颌骨硬化加重,锁骨和肋骨宽度和密度增大。患者全身皮质增生、外部轮廓轻微改变、听力正常和颅内压。Itin等人黑色箭头表示导致CLS并指的突变位点,紫色箭头表示导致CMS17先天性肌无力的突变位点,蓝色箭头表示导致2型硬化性骨病的突变位点。报道了一名36岁的希腊妇女患有脑神经疾病,包括双侧周围性面瘫、听力损失和脑神经孔增生变窄。此外,双侧食指发育不良和指甲发育不良分为两部分,尤其是食指。影像学检查显示颅骨骨质增生,手脚骨皮质增厚。
[24]
 [25]
[25]
[26]
人类骨矿化过程和由LRP4功能障碍引起的异常骨量调节被诊断为2型硬化性骨病。Leupin等人[8]通过对2型硬化性骨病患者的两个家族进行基因筛查,最终确定该疾病的致病基因为LRP4,并鉴定出两个全新的LRP4错义点突变。体外实验证实,两种突变体LRP4都能正常转移到细胞膜上。他们还发现LRP4与SOST蛋白(硬化症)相互作用。SOST是1型硬化性骨病的致病基因,也是Wnt信号通路的抑制剂。硅核糖核酸敲除LRP4并过度表达LRP4。发现LRP4与WNT信号通路相关,SOST可能通过LRP4 [8对经典Wnt信号通路进行负调节。此后,Fijalkowski等人[27]报道了一个新的导致2型硬化性骨病的LRP4点突变,并发现所有报道的导致2型硬化性骨病的LRP4突变蛋白都不能拮抗LRP5激活的经典Wnt信号通路,这与导致CLS并指的LRP4突变蛋白一致。熊等人2.3 LRP4和CMS17重症肌无力发现,在破骨细胞特异性敲除Lrp4的小鼠中,突变小鼠的骨吸收受到抑制,并且在突变小鼠模型中,皮质和骨小梁的骨量增加,骨吸收受损,但是没有并指表型和神经发育的异常。这与其他Lrp4缺陷小鼠非常不同。Boudin等人CMS 17是一种常染色体隐性遗传疾病,一种先天性重症肌无力,由Ohkawara等人在2014年首次报道[[9]。一名年轻女性患者自出生以来呼吸和进食困难,仅在18个月后才学会走路,并且容易疲劳。她童年的一部分时间是在轮椅上度过的。9岁和14岁时的检查显示,患者的上眼睑下垂,眼球横向运动受到轻微限制,中度至重度远端无力和反射不足。重复神经刺激对快速作用的胆碱受体激动剂仅表现出微弱的反应。骨骼肌活检显示1型纤维排列不规则,使得突触连接的形状和大小非常不同。电镜结果显示,神经末梢大小降至60%,突触后区大小降至48%。乙酰胆碱受体和乙酰胆碱酯酶的表达是正常的[9]。最近的一项研究发现,通过基因敲入技术,小鼠LRP4基因的1170精氨酸突变为谷氨酰胺(Lrp4点突变导致硬化性骨病ⅱ型),筛选出Lrp41170Q敲入纯合的突变小鼠。体内实验证实,Lrp4-R170q点突变确实影响骨矿化和骨吸收过程,小鼠表现出硬化性骨病表型,突变小鼠的肢端和神经肌肉接头发育正常。这表明该位点的突变确实影响骨矿化和骨量调节过程,表明LRP4确实是2型硬化性骨病的致病基因。导致2型硬化性骨病的LRP4突变位点和突变类型在图1中用蓝色箭头表示。
[28]
[29]
LRP4参与神经肌肉接头发育的调节。在人类中,由LRP4功能障碍引起的神经肌肉接头发育不良被诊断为CMS17肌无力综合征。Ohkawara等人,[9]在2014年首次报道了这种单一基因疾病。对患者家族的主要家族成员进行全外显子测序并结合第一代测序基因筛选,最终确定该疾病的致病基因为LRP4。鉴定出两个全新的LRP4点突变,位于LRP4的第三个β螺旋结构域,这对LRP4/MUSK/AGRIN复合物的形成非常重要(AGRIN是一种源自运动神经元的配体,可以激活MUSK磷酸化,MUSK是一种在骨骼肌中表达的受体酪氨酸激酶)。目前,只报道了一个家族,导致如图1中紫色箭头标记所示的CMS17的LRP4突变位点和突变类型。根据双荧光素酶实验,野生型Lrp4蛋白可以激活ATF2介导的JNK信号通路(MUSK激活的下游基因,可以取代MUSK作为检测指标),但两种Lrp4错义点突变蛋白导致CMS17失去激活JNK信号通路的能力,但仍然可以拮抗经典Wnt信号通路;Lrp4错义点突变蛋白导致CLS并指,能激活JNK信号通路,不能拮抗经典Wnt信号通路[9]。韦瑟比等人[20]发现,两只ENU筛选的点突变小鼠,Lrp4mte和Lrp4mitt,除肢端肥大症外,不能存活。经分析检测,发现突变小鼠神经肌肉接头处突触后细胞乙酰胆碱受体的聚集受到抑制,导致窒息和过早死亡。Kim等人[30]和Zhang等人3 LRP4点突变和3种罕见遗传疾病通过体外实验证实,Lrp4是Agrin(小鼠ARGIN同源基因)的一种新的共受体,它可以与Musk(小鼠MUSK同源基因)和Agrin结合形成一种复合体来调节突触后转化。Lrp4的缺乏将导致麝香磷酸化过程的抑制,这将抑制突触后膜中乙酰胆碱受体(ACh R)的聚集,并阻止突触后转化过程的激活。Yumoto等人LRP4的不同点突变可以导致完全不同的疾病(图1)。根据关于LRP4的报道,CLS型LRP4点突变占优势,共有14个,分布在LRP4的胞外区,相对分散。李等人[7]发现CLS型LRP4点突变不能拮抗经典Wnt信号通路,转位到细胞膜的过程受到抑制。异常易位可能导致LRP4失去某些功能,无法对信号网络做出反应,导致肢体和肾脏发育异常。2型硬皮病LRP4有3个点突变,都位于第三个β螺旋结构域。Leupin等人[8]发现,导致2型硬化性骨病的LRP4点突变可以被正确靶向并转运至细胞膜,但其与SOST的结合被削弱,并且不能拮抗经典Wnt信号通路。据推测,这种结合的减弱不能拮抗经典Wnt途径,加速骨硬化和骨矿化过程,并导致骨硬化。CMS17型LRP4有2个点突变,都位于第三个β螺旋结构域。Ohkawara等人[9]发现,该区域的氨基酸变化将失去激活JNK途径的能力,并可靶向细胞膜。该区域对LRP4/MUSK/AGRIN复合体的形成非常重要。因此,该区域的两个点突变可能影响LRP4/MUSK/AGRIN复合体的形成,从而影响NMJ的正常发育并导致CMS17重症肌无力。证实,Lrp4可以通过小鼠体内实验与Agrin结合,激活麝香并促进突触后转化。此外,Lrp4也可用作肌源性逆行信号,这是突触前转化的必要和充分的信号。
[31]
[32]
4 LRP4调节信号通路和相关基因
Johnson等人[6]和Ohkawara等人[9]揭示了Lrp4可以通过体外双荧光素酶实验拮抗经典Wnt并激活JNK信号通路。后来,李等人[7]和菲贾科夫斯基等人[27]发现导致CLS并指和2型硬化性骨病的Lp4突变蛋白不能拮抗经典Wnt信号通路,导致CMS17的Lp4突变蛋白失去激活JNK信号通路的能力。经典Wnt信号通路具有多种生物学功能,参与调节细胞生长、迁移和分化,调节正常组织重建和其他生命活动,并在出生后胚胎和骨发育形成中的肢体和骨发育中发挥重要作用[33]。目前,据信LRP4在肢体发育过程中通过竞争性结合Wnt配体与LRP5/LRP6对抗经典WNT信号通路。如果LRP4缺失,将导致肢体发育障碍。在成骨细胞和破骨细胞中,SOST通过LRP4拮抗经典Wnt信号通路,而LRP4的减少将导致过度骨生长和骨硬化[7,8]。JNK信号通路家族是丝裂原活化蛋白激酶超家族的成员之一。它在基因表达、细胞增殖、细胞应激、细胞凋亡促进等许多生理和病理过程中发挥重要作用。信号通路与神经系统损伤的发生和修复密切相关,[34]。LRP4可以激活JNK信号通路。如果神经肌肉接头中LRP4减少,MUSK磷酸化被抑制,突触后转化不能被激活,骨骼不能感知上游刺激,将产生肌无力表型(图2)。
Johnson等人[6]通过在Lrp4ECD/ECD小鼠胚胎上的原位杂交实验,发现Fgf 8(小鼠成纤维细胞生长因子8编码基因)、Shh(参与早期胚胎轴发育的重要蛋白编码基因)、Bmp2(小鼠骨形态发生蛋白2编码基因)、Bmp4(小鼠骨形态发生蛋白4编码基因)和Wnt7a(小鼠Wnt家族成员7a编码基因)基因的异位表达。Asai等人[35]在小鼠ATDC5细胞系中构建Sh核糖核酸以连续敲除Lrp4,并发现软骨标记基因Col2a1(小鼠ⅱ型胶原α1链编码基因)、Acan(小鼠聚集蛋白聚糖编码基因)和Col10a1(小鼠10型胶原α1链编码基因)的表达降低。成骨标记基因Sox9(小鼠性别决定区Y盒9蛋白编码基因)、Runx2(小鼠RUNT相关转录因子2编码基因)、Mmp13(小鼠基质金属蛋白酶13编码基因)、Dkk1(小鼠Wnt信号通路抑制蛋白Dickkopf 1编码基因)和Sost(小鼠硬化蛋白编码基因)的表达显著降低。Wnt信号通路Wnt3a(小鼠Wnt家族成员3a的编码基因)和Axin2(小鼠axin2的编码基因)显著上调。LRP4的主要功能是参与经典Wnt信号通路和JNK信号通路的调节,这可能与其他信号通路有关,但目前还没有实质性的证据。
目前发现的LRP4配体主要有:Agrin、Sost、DKK1、Gremlin1(小鼠DAN家族Bmp信号通路拮抗蛋白)、Wise(小鼠硬化域含蛋白)、Wnt、APP(小鼠淀粉样β-A4蛋白)和Apo E(小鼠载脂蛋白E)。LRP4和AGRIN形成的综合体对NMJ的发展和功能维护至关重要。SOST和DKK1与LRP4的结合能负调节经典的WNT信号通路,LRP4和Wnt蛋白的竞争降低LRP5/LRP6的结合效率,从而拮抗经典的Wnt信号通路。LRP4和Wise的组合调节Wnt信号通路,促进表皮细胞分化和乳房发育。LRP4和Gremlin1可以相互作用,并且可以间接调节骨形态发生蛋白信号通路[36]。
5结论和前景
虽然LRP4对脊椎动物早期发育的分子调控机制尚不十分清楚,但多年的体内和体外研究已经证明,Lrp4缺陷小鼠在肢体、肾脏、神经肌肉接头、乳房和牙齿方面存在发育异常,并且骨矿化和骨量调节受到影响。
目前,LRP4调节神经肌肉接头发育和骨代谢过程的调节机制已逐渐阐明。LRP4和AGRIN/MUSK形成一个复合体来促进突触后转化和调节神经肌肉接头的信号传递。在成骨细胞和破骨细胞中,SOST通过LRP4调节经典Wnt介导的骨量增加和减少,以维持骨盐平衡。脊椎动物肢体发育是一个高度协调、复杂和精确的调节过程。目前,对LRP4调节肢体发育的分子机制知之甚少。只有一个经典的Wnt信号通路调节肢体发育。无论是Lp4缺陷小鼠还是CLS并指患者,肢体表型都非常严重,掌骨、腕骨、桡骨和尺骨都严重紊乱。它与前人报道的经典Wnt途径缺陷小鼠的表型不一致,更严重。此外,一条信号通路同时导致两种不同的疾病,这很难让人信服。因此,有必要进一步探索LRP4的潜在反应途径。
图2 LRP 4参与的信号通路及不同组织器官的内部调节机制
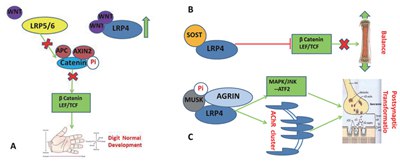
答:LRP4参与肢体发育调控;骨吸收蛋白4参与调节骨量的增加和减少;C:LRP4参与神经肌肉接头的信号传递
在由报道的LRP4突变引起的三种疾病中,表型是单一的并且不相互干扰。CLS肢体异常患者的神经肌肉接头发育和骨代谢正常。另外两种疾病相似。因此,我们推测LRP4在不同组织和器官的发育调控中可能具有不同的相互作用蛋白和信号网络。当LRP4经历影响肢体发育的错义点突变时,它仅部分失去功能,并能继续调节其他组织和器官的正常发育。Lrp4的分子机制有待进一步研究。基因敲入技术可能成为研究这种多功能基因的新选择。Lrp41170Q基因敲入的点突变小鼠非常全面地模拟了2型硬化性骨病的表型,其他指标也表明肢端和神经肌肉接头的发育不受影响。在未来多功能基因的功能验证中,基因敲入可能是一个新的视角。
进一步研究LRP4有助于进一步了解肢体发育过程,筛选出CLS并指或诱导并指等肢体发育异常的其他候选基因,为临床诊断和准确治疗提供理论依据。
参考
[1]李英,金军,卜红。低密度脂蛋白受体家族:细胞内吞和信号转导。Mol神经iol,2001,23:53-67
[[2]沈c,熊维康,梅丽尔。LRP4在神经肌肉接头和骨发育及疾病中的作用。骨骼,2015,80:101-8
[3]平森基,布伦南J,蒙克利S,等。一种低密度脂蛋白受体相关蛋白介导小鼠Wnt信号。自然,2000,407:535-8
[4]田美君,塞门诺夫,加藤,等. Wnt信号转导中的低密度脂蛋白受体相关蛋白。自然,2000,407:530-5
[5]Nakayama M,Nakajima D,Nagase T,等.通过模体陷阱筛选鉴定具有多个表皮生长因子样模体的高分子量蛋白质。基因组学,1998,51:27-34
[6]约翰逊·EB,哈默·雷,赫茨·J .兆福缺陷小鼠顶外胚层嵴和多指畸形发育。哼摩尔热奈特,2005,14:3523-38
[7]李勇,鲍利克,埃尔乔古鲁,等。LRP4突变改变Wnt/β-连环蛋白信号,并导致塞纳-伦茨综合征的肢体和肾脏畸形。《基因杂志》,2010,86:696-706
[8]亮蛋白0,皮特尔斯E,哈勒克斯C,等。骨过度生长与LRP4基因突变相关,损害硬化素促进因子功能。《生物化学》,2011,286:19489-500
[9]Ohkawara B,Cabrera-Serrano M,Nakata T,等. LRP 4thirdβ-螺旋桨结构域突变通过以位置特异性方式损害农业蛋白介导的Mu SK信号而导致新的先天性肌无力。哼摩尔热奈特,2014,23:1856-68
[10]塞纳尼,伦茨·威。2兄弟中的全并指和全桡尺滑膜。syndactylia的遗传学研究进展。Z Kinderheilkd,1967,101:181-90
[11]利伯曼·莱博·格雷切蒂格·沃科曼·冯·格列德马斯恩德费肯和骨肉瘤治疗系统。z menschliche Vererbungs-und Koinstitutionslhere,1938,21:697-70
[12]博尔斯基AJ。先天性手部异常及其外科治疗。《儿科学》,1958,53:759
[13]叶尔顿CL。双胞胎和同父异母兄弟姐妹的某些先天性肢体缺陷。诊所间通知公牛,1962,1:1-7
[14]汗TN,克拉尔J,阿里Z,等。塞纳尼-伦兹综合征限于肢体和肾脏异常相关的一个新的LRP4错义突变。《欧洲医学遗传学》,2013,56:371-4
[15]卡里姆尼贾德·阿,斯托尔夫?李勇,等。因LRP4功能丧失引起的重度森尼林综合征。医学遗传学杂志,2013,161A:1475-9
[16]呼唤AS,布普CP,Mc Gee SJ,等。截短LRP4的突变导致产前致死形式的塞纳-伦茨综合征。医学遗传学杂志,2014,164A:2391-7
[17]阿夫扎尔·米,扎曼·克,库尔纳克·乌,等。LRP4中的新剪接突变导致严重类型的塞纳-伦茨并指综合征,伴有口面部和骨骼症状。《欧洲医学遗传学》,2017,60:421-5
[18]哈普夫·C,帕维尔卡·M,胡塞尔·H .塞纳尼-伦茨辛德提(CLS):文献回顾与分类尝试。《塑料外科》,2005,58:251-7
[19]西蒙-查佐特斯·德,图图瓦·斯,库恩·姆,等。编码低密度脂蛋白受体LRP4的基因突变导致小鼠肢体发育异常。基因组学,2006,87:673-7
[20]威瑟比SD,安德森千伏,尼斯文德洛杉矶。乳酸脱氢酶受体相关蛋白4对神经肌肉接头的形成至关重要。发展,2006,133:4993-5000
[21]安·Y,西姆斯·C,罗格·JM等。Lrp4和怀斯相互作用通过调节Wnt信号来控制乳腺和其他皮肤附件板的形成和模式。发展,2013,140:583-93
[22]约翰逊·EB,斯特芬·DJ,林奇·KW,等.常染色体隐性多足病中远端肢体发育的调节因子Megf7/Lrp4的缺陷剪接.基因组学,2006,88:600-9
[23]卡尔纳·CM,迪特里希·MF,约翰逊·EB,等。LRP 4调节输尿管出芽的开始,并对肾脏形成至关重要——肾尼-伦茨综合征的小鼠模型。PlO S1,2010,5:e10418
[24]Tanahashi H,Tian QB,Hara Y,等. Lrp4基因敲除小鼠双侧肾脏发育不全时羊水过多:羊水清除途径的缺陷.Sci代表,2016,6:20241
[25]布埃诺·姆,奥利万·格,希门尼斯·阿,等。西班牙男性的硬化症:地中海裔人的首次报告。《医学遗传学》,1994,31:976-7
[26]将其作为硬化症的标志病变。皮肤病学,2001,202:259-60
[27]菲贾科夫斯基ⅰ,吉茨E,斯蒂纳克尔斯E,等。硬化症患者中的一个新的结构域特异性突变表明LRP4作为人类骨骼中硬化素的锚定物的作用。《骨矿资源》,2016年,31:874-81
[28]熊磊,郑菊,吴虎,等.成骨细胞中的Lrp4抑制骨形成,促进破骨细胞形成和骨吸收。美国国家科学院学报,2015,112:3487-92
[29]鲍丁·E,约尔根·T,菲贾科夫斯基等人。Lrp4R1170Q纯合敲入小鼠概括了人类硬化症的骨表型。《骨矿资源》,2017年,32:1739-49
[30]金·恩(Kim N),斯蒂格勒·艾尔(Stiegler AL),卡梅伦·托(Cameron TO)等人,Lrp4是阿格林的受体,与麝香形成复合体。手机,2008,135:334-42
[31]张博,罗思,王问,等。LRP4作为农业生产者的共同接受者。神经元,2008,60:285-97
[32]玉本,金恩,负担SJ。Lrp4是神经肌肉突触突触突触前分化的逆行信号。自然,2012,489:438-42
[33]麦KK,陈厚海,戴TF,等al.Wnt/β-catenin信号在控制软骨内骨和滑膜关节形成中与Ihh信号有差异地相互作用。发展,2006,133:3695-707
神经退行性疾病:从现有疗法到未来草药疗法。神经化学国际,2016,95:100-8
[35]Asai N,Ohkawara B,Ito M,等. LRP4诱导细胞外基质产生并促进软骨细胞分化。生物化学生物物理学通讯,2014,451:302-7
[36]蔡海英,刘Y,丁耐特,等。APP与LRP 4和agrin相互作用,协调小鼠神经肌肉接头的发育。Elife,2013,2:e00220
关于遗传疾病的第一篇论文(2)
主题:基因诊断技术在出生缺陷和遗传病检测领域的应用研究
摘要:目的分析基因诊断技术在出生缺陷和遗传病检测领域的应用效果。方法以89例因出生缺陷和遗传病就诊后确诊的产妇为研究对象。所有产妇均接受超声检查和基因诊断,并比较两种方法的诊断结果。结果超声检查发现异常胎儿67例,确诊率75.28%,漏诊率24.72%,诊断符合率98.51%。基因诊断检出异常胎儿82例,诊断率92.13%,漏诊率7.87%,诊断符合率100.00%。两种方法的差异有统计学意义(P0.05)。结论基因诊断在出生缺陷和遗传病检测中比超声有更高的应用价值,能更准确地诊断疾病。然而,这两种检测技术的侧重点不同,可以结合使用,提高出生缺陷和遗传病患儿的诊断率。
关键词:出生缺陷;遗传病;基因诊断技术;超声波诊断;
基因诊断技术在出生缺陷和遗传病检测领域的应用研究
李英昌黄绍罗真肖娟
佛山市第一人民医院
摘要:
目的分析基因诊断技术在出生缺陷和遗传病检测领域的应用效果。方法选择89例经咨询确诊为出生缺陷和遗传病的孕妇作为研究对象。所有产妇均接受超声和基因诊断检查。比较了两种方法的诊断结果。结果超声检查发现异常胎儿67例,准确率为75.28%,漏诊率为24.72%,诊断符合率为98.51%。基因诊断发现异常胎儿82例。诊断率为92.13%,漏诊率为7.87%,诊断符合率为100.00%。两种方法在诊断率上有显著差异(P0.05)。结论基因诊断在出生缺陷和遗传病的检测中比超声诊断更有价值,能更准确地诊断疾病。然而,这两种检测技术有不同的侧重点,可以结合使用,以提高出生缺陷和遗传病的诊断率。
关键词:
出生缺陷;遗传病;遗传诊断技术;超声波诊断;
出生缺陷是胎儿畸形,是指发生在母亲体内的结构或染色体异常。根据调查和统计,畸形胎儿占活产的3%。世界上每年新增约500万个畸形胎儿,主要是在发展中国家。中国先天性残疾儿童的发病率高达80万-120万,占出生总数的4%-6%。其中大多数是由遗传、环境和综合因素造成的,这些因素不仅导致胎儿残疾,还增加了围产期死亡的风险。遗传病是指由遗传物质或致病基因的变化引起的疾病。它们的特点是垂直传播和终身性。先天性疾病和获得性疾病有两种类型。临床常见疾病包括先天性愚蠢、先天性耳聋、多趾症和血友病,这些疾病严重影响了他们的生活质量和未来的生长发育[1]。随着医疗卫生事业的进步,产前筛查技术越来越完善,可以尽早诊断异常胚胎,为后续医疗工作提供参考。目前,产前筛查技术越来越多样化。如何科学地使用它来提高诊断的准确性,已经成为医生和病人关注的焦点[2]。本文研究了基因诊断技术在出生缺陷和遗传病检测领域的应用价值。报告如下。
1数据和方法
1.1一般信息
选取2015年1月至2017年1月在我院就诊的89例出生缺陷和遗传病产妇作为研究对象。所有产妇都同意参与这项研究,排除那些患有精神交流障碍的产妇。这项研究得到了医学伦理委员会的批准。产妇平均年龄为(29.64±3.15)岁。平均孕周为(20.32±1.15)周。其中,产褥期生畸形儿7例,孕期服药8例,血型不合夫妇5例。
1.2研究方法
所有产妇都接受了超声波检查和基因诊断。检查由同一医疗小组和2-3名医生进行,以确保检查的科学性。(1)超声检查:选用美国通用电气公司的通用电气公司的v730四维彩色多普勒超声仪对产妇进行腹部检查。检查时产妇保持仰卧位,在腹部涂抹消毒超声耦合剂,并严格检查腹内胎儿、胎盘和羊水情况。护士记录并整理检查数据,供专业医生诊断。(2)基因诊断:医院选择罗氏实时荧光定量聚合酶链反应(聚合酶链反应)仪进行检查,并从晨间空腹部组织产妇血液采样进行严格检测,科学分析产妇外周血清和血浆中游离胎儿脱氧核糖核酸,分析产妇性染色体和血液中染色体,整理检测数据供医生诊断。
1.3观察指标
根据分娩情况,对诊断结果进行分析,比较超声检查和基因诊断在出生缺陷和遗传病检测中的诊断率、漏诊率和诊断符合率。
1.4统计方法
采用SPSS20.0统计学软件对数据进行统计分析。计量资料以均数±标准差 (?±s) 表示, 采用t检验;计数资料以率 (%) 表示, 采用χ2检验。P


