性发育异常疾病的分类和临床表现研究,软骨终板多发性发育不良是什么疾病
性发育异常疾病的分类和临床表现研究
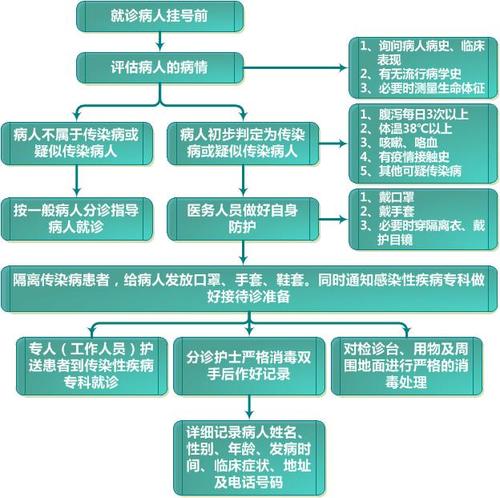
软骨终板多发性发育不良的疾病到底是什么?中药可用于椎体终板骨软骨炎的对症治疗。建议:关键是排除椎间盘突出症等疾病,因为左臀部放射痛是腰椎间盘突出症最典型的症状。因此,应该将其排除在外。如果是腰椎间盘突出症,需要绝对卧床休息,症状无法缓解。建议去看医院,因为这种疾病可能需要
胸腺发育异常属于获得性免疫缺陷病吗
它不是获得性免疫缺陷病,而是先天性免疫缺陷性玻璃体胸腺发育不全,别名先天性胸腺发育不全,也称为DiGeorge异常或ⅲ、ⅳ咽囊综合征。 它可以以常染色体显性或隐性遗传为特征。 该疾病是由先天性胸腺发育不良或发育不良引起的一种T细胞功能障碍疾病。细胞:(1)下丘脑闭经下丘脑闭经是由各种下丘脑功能和器质性疾病引起的闭经 这种闭经的特点是垂体促性腺激素(Gn)分泌不足,即卵泡刺激素(FSH)和黄体生成素(LH),特别是黄体生成素(LH),由下丘脑合成和促性腺激素释放激素(GnRH)分泌不足或不足引起。我不知道你具体指的是哪个方面,是指身体、智力还是其他方面。智力有很大的遗传关系,身体发育主要受内分泌系统的影响。男孩男性激素分泌不足,但男性特征不明显,阴茎短小,声音尖细,而女孩磁性激素分泌不足,声音粗糙,会导致心肌缺血、心肌炎和头发过多等心肌损伤。 也有一些功能性ST-T改变,这是由自主神经功能的改变引起的。 几乎所有先天性获得性心脏病都是可以引起的。自主神经也可能引起。一些心脏外疾病也可能导致。 牙齿发育异常包括双齿畸形、先天性缺牙、乳阿桔、牙釉质发育不全、上前牙异常尖、乳牙滞留、乳牙粘连、中位牙、多生牙、牙齿大小异常(过大牙、过小牙)等。 它们会导致乳牙脱位和乳牙列固有长度的缩短,导致遗传恒牙排列拥挤。 例如下颌乳前牙的固位,
软骨终板多发性发育不良是什么疾病
软骨终板多发性发育不良的疾病到底是什么?中药可用于椎体终板骨软骨炎的对症治疗。建议:关键是排除椎间盘突出症等疾病,因为左臀部放射痛是腰椎间盘突出症最典型的症状。因此,应该将其排除在外。如果是腰椎间盘突出症,需要绝对卧床休息,症状无法缓解。建议去看医院,因为这种疾病可能需要
胸腺发育异常属于获得性免疫缺陷病吗
性发育异常疾病的分类和临床表现研究范文
摘要:性腺和性发育是一个连续有序的过程。正常性腺的发育主要依赖于两个因素:染色体(XY,XX)和调控胎儿宫内性腺形成、分化和发育的相关因素。一旦这一过程中出现异常,就会导致性发育不良疾病的发生。本文综述了性发育不良的定义、性发育的遗传基础、常见性发育不良疾病的分类和临床表现,重点介绍了几个相关致病基因。
关键词:性发育不良;性别决定;性别分化;基因突变;
性发育异常(Disorders of sexual development,DSD),又称性分化异常,是在性别决定和分化过程中,由染色体畸变或基因突变引起的性发育和内分泌调节异常,包括性染色体异常、性腺发育异常、副性器官解剖异常等。
1人类性别决定和性别分化
性发育是一个复杂而持续的过程,包括从原始性腺形成睾丸或卵巢(性别决定)和内外生殖器的分化(性别分化)。首先,不同的性染色体(XX或XY)产生自己的性腺(睾丸或卵巢),通过相关激素的调节,它们分化成不同的性生殖管道和外生殖器。
1.1性别决定
胚胎原始性腺的发育命运由性染色体的组成决定。一般来说,含有γ的精子和卵子是受精的。合子染色体是46,XY。原始性腺发育成睾丸并变成雄性。精子和含x的卵子受精,合子染色体为46,XX,原始性腺为卵巢,发育为雌性。
1.2性别差异
人性腺原基出现在妊娠第4周的中胚层。在分化为睾丸或卵巢之前,它将经历一个未分化期。此时,性腺没有任何性别特征。睾丸在早期胚胎中分泌睾丸素和AMH,睾丸素转化为二氢睾丸素,使内外生殖器发育成男性。没有睾酮和AMH,肾旁管发育成女性内生殖器和外生殖器,所以女性生殖器的发育不依赖卵巢,而是从属于性腺的分化。
青春期后,男性和女性生殖器官逐渐成熟。男性青春期通常在14到20岁之间。青春期开始后,生精小管迅速生长,睾丸体积明显增大,附睾、精囊和前列腺也逐渐成熟,阴囊皮肤变得松弛和着色,阴毛和腋毛开始出现,随后胡须、喉结和嗓音发生变化。女孩比男孩早两年开始青春。月经初潮是性功能发展的主要标志。乳房发育、大阴唇和小阴唇发育、色素沉着、阴道分泌物增多、阴毛和腋毛以及卵巢、输卵管、子宫和阴道逐渐发育成熟。
2常见性发育不良疾病的分类和临床表现
根据DSD的新分类,它可以分为三种类型:性染色体DSD,46,XY DSD和46,XX DSD。性染色体DSD患者有特殊的核型。46,XY DSD和46,XX DSD涉及多个基因,它们在许多方面是如性连锁遗传、常染色体显性遗传和常染色体隐性遗传。
目前对基因控制发育的理解主要来源于与糖尿病患者相关的基因突变和功能分析。小鼠模型和DSD体外模型(如人卵巢畸胎瘤细胞、人多能干细胞睾丸胚胎癌细胞系等。)已被广泛应用于[1】。在哺乳动物中,性发育的机制可以在老鼠身上得到彻底的研究和最好的理解。性发育是一个涉及大量基因的复杂过程。常见糖尿病的遗传学和发病机制以及该疾病的相关小鼠模型见表1-3。
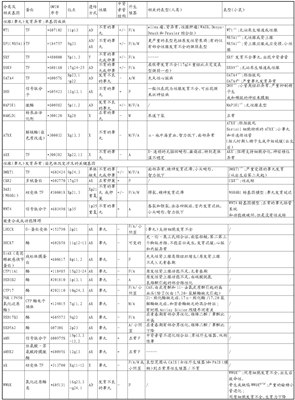
2.1性染色体DSD的主要类型和临床表现见表1。
表1性染色体DSD的主要类型和临床表现

外宾:像女人一样;男:像男人一样;性别模糊
2.2 46,XY DSD
也被称为睾丸发育不良,其主要类型、涉及的基因、临床表现和小鼠模型见表2。目前,小鼠模型主要用于以下基因突变。
(1) ARX
同源框蛋白ARX是一种转录因子,在小鼠胚胎发育过程中在前脑、底板和睾丸中表达。在人类中,ARX基因主要在胎儿大脑、成人心脏、骨骼肌和肝脏中表达。ARX基因敲除小鼠在胚胎发育过程中表现出异常的神经元迁移和阻碍睾丸间质细胞分化(性腺发育不全),这将导致睾丸变小、生精小管大量和精囊发育不全。在人类中,ARX突变发生在患有X连锁无脑回和不明生殖器综合征的患者中,[2]。
(2) ATRX
ATR-X综合征是由ATRX突变引起的一种与染色体相关的疾病。这种疾病的特征是严重的精神发育迟滞、典型的面部特征、睾丸小且没有或只有少量生殖细胞、生殖器异常和α地中海贫血[3,4]。ATRX基因敲除小鼠在世界范围内对胚胎是致命的。ATRX是一种染色质重塑蛋白,作为共激活剂直接与雄激素受体相互作用,[5]。
(3) CBX2
在雌性和雄性Cbx2 (M33)敲除小鼠中,性腺发育不良出现,而XY Cbx2敲除小鼠显示性别逆转。XY Cbx2敲除小鼠性腺中,SRY和SOX9基因表达降低。在人类中,在卵巢组织学正常且女性内外生殖器正常的46例XY DSD患者中鉴定出CBX2复合杂合突变。
(4) DHH
被DHH基因敲除的XY小鼠不育,管周组织异常,严重抑制精子发生[6】。在神经系统中,神经束膜周围的周围神经簇异常薄。这种性腺表型表明,DHH在肌肉和睾丸间质细胞发育过程中同时作用于它们。人DHH突变是常染色体隐性突变,发生在46例完全或部分性腺发育不全(伴或不伴微泡神经病变)的XY患者中。在人类中,这两名患者患有混合性性腺发育不全,其特征是一侧睾丸和对侧DHH索状杂合突变(性腺发育不全是两性的典型特征)或另一侧没有性腺[7]。
(5) DMRT1
在人类中,DMRT1在XY的胚胎生殖嵴(但不是XX)中表达。性腺发育不全的46,XY DSD与单体9 p末端相关,该染色体区域含有DMRT1。DMRT1外显子3-4缺失在46,XY oorchid DSD患者[8]中被鉴定。相比之下,Dmrt1敲除小鼠从出生后第二天起表现出严重的睾丸发育障碍,导致严重的睾丸畸形,但没有性逆转。
(6) GATA4
人类GATA4突变与先天性心脏病有关。此外,在一个有46,XY DSD和先天性心脏病[9]的家庭成员中报道了GATA4的杂合错义突变。虽然Gata4基因敲除小鼠在性腺分化前死于胚胎,Gata4 Val271Gly突变小鼠在性交后存活13.5天,显示睾丸发育不良,表明GATA-4和FOG-2之间的相互作用对于SRY基因的正常表达是必需的,这解释了正常人性腺中的表型[10]。
(7)MAMD1
在患有由MAMLD1突变引起的单纯性尿道下裂的个体中。在小鼠中,Mamld1在胎儿滋养层和睾丸间质细胞中瞬时表达。细胞研究表明,马姆尔德1的短期敲除降低了SF-1介导的Cyp17a1表达,从而抑制睾酮的产生。因此,MAMLD1突变患者的生殖器表型可能是由于胚胎发生关键时期短暂的睾丸功能障碍导致睾酮产生的损害。
(8) MAP3K1
两个家族和两个46,XY DSD性腺发育不良患者之间的连锁分析显示MAP3K1基因有4个杂合突变。MAP3K 3在胚胎小鼠的性腺中表达,但是没有MAP3K1的小鼠是可育的,并且不显示性腺异常。虽然MAPK信号在睾丸发育中很重要,但人类和小鼠可能会利用MAP3K家族成员[的不同作用。人类MAP3K1获得的功能突变是否会干扰睾丸前支持细胞的分化尚不清楚。
(9) NR0B1
NR0B1被认为是抗睾丸基因,因为人类染色体Xp21区域(包括NR0B1)的复制将导致46,XY性腺发育不良。然而,过表达Nr0b1的XY小鼠在弱SRY基因表达背景下会表现出性别逆转,表明DAX-1(蛋白产物Nr0b1)对性别发育具有剂量敏感效应。
(10) NR5A1
NR5A1在泌尿生殖嵴中表达,其基因产物为SF-1,激活SRY基因表达。NR5A1基因敲除小鼠缺乏肾上腺和性腺,出生后8天死于肾上腺功能不全。在人类中,NR5A1的杂合突变(很少是纯合的NR5A1突变)发生在性腺发育不全和新生儿肾上腺衰竭的患者中46,XY DSD [12]。
(11) WT1
WT1是锌指转录因子,在生殖嵴、胎儿性腺和发育中的肾脏中表达。WT1敲除小鼠的肾脏和性腺发育受阻,导致胚胎致死率。
(12) WWOX
在[13]的46例XY DSD和性腺发育不全患者中发现了从母体来源获得的WWOX等位基因外显子6-8的杂合缺失。完全WWOX敲除小鼠在3周龄时死亡,性腺表型为雌雄同体。
2.3 46,XX每日生活津贴
也被称为卵巢发育障碍,其主要类型、涉及的基因、临床表现和小鼠模型见表3。目前,小鼠模型主要用于以下基因突变:
(1)MAMD1
性腺发育不全的46,XX DSD MAMLD1基因具有纯合错义功能获得性突变。在另一个患有性腺发育不全的46位XY DSD患者中,它是在同一位点的杂合突变,表明MAMLD1编码的蛋白质可能参与睾丸发育[14]。
(2) NR5A1
颗粒细胞特异性XX NR5A1-/-小鼠因卵巢发育不良、排卵障碍和不孕而未达到性成熟,[15]。在对139例原发性卵巢衰竭患者的实验中,7名[人发现CITED2 (NR5A1上游调节因子)突变。
(3) RSPO1
在人类中,46,20个睾丸DSD家族报告了RSPO1纯合突变,伴有手掌和脚底角化过度和皮肤鳞状细胞癌,[17]。随后,报告了46例卵巢囊肿型糖尿病。响应素-1通过(可能协同地)通过WNT4基因[18,19促进卵巢颗粒细胞的分化来增强β-连环蛋白的信号。被Rspo1击倒的二十只小鼠没有表现出完全的性别逆转。然而,它们有男性卵巢(胎儿卵母细胞耗尽)、男性特有的体腔血管结构和异常睾酮合成。
(4) SOX3
SRY相关的SOX-3基因在人和小鼠的性腺发育中非常低或不表达。SOX3功能突变的缺失不会影响人类和小鼠的性别发育,[20]。然而,在双潜能性腺中过量表达SOX3的转基因品系××小鼠表现出完全的性别逆转。
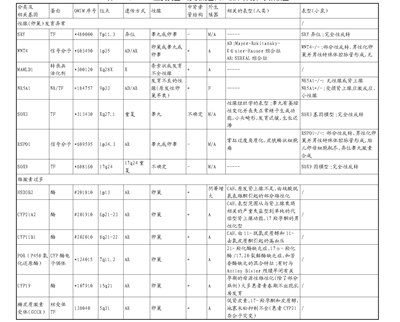
表2 46,XY DSD的主要类型、相关基因、临床表现和小鼠模型
转录因子;核受体;广告:常染色体显性;AR:常染色体隐性遗传;十:十染色体连锁;Y:Y染色体连锁;CAH:肾上腺皮质增生;血清激肽释放酶综合征(SERKAL syndrome):由WNT4基因突变引起的常染色体遗传病,功能丧失;WAGR:威尔曼肿瘤、无虹膜、泌尿生殖系统异常和精神发育迟滞综合征;梅耶尔-罗基坦斯基-卡斯特-豪泽综合征:先天性阴道缺失和部分阴道缺损
表3 46,xDSD的主要类型、相关基因、临床表现和小鼠模型
(5) SOX9
SOX9在XX小鼠性腺中的过表达导致睾丸的形成,小鼠的表型为雄性。
(6) SRY
在人类中,超过80%的患有46,20个睾丸dsd的患者在X染色体上检测到SRY易位。此外,过表达SRY的二十只转基因小鼠发育成雄性,证明了SRY是睾丸发育唯一必需和足够的Y染色体基因。
(7) WNT4
在小鼠胚胎中,WNT4在交配后11天在雄性和雌性的双潜能性腺基质细胞中表达。基因敲除的二十只小鼠卵巢呈阳性,缺少中肾。虽然没有WNT4(纯合敲除)的小鼠在出生后死于肾衰竭,但是只有一部分WNT4杂合人群患有肾功能障碍。在人类患者中,WNT4基因缺失的小鼠出生时没有中肾,也没有子宫或发育不良。
2.4多基因dsd
对多基因DSD患者的诊断可能会发现由复杂遗传异常引起的新型DSD。例如,一名患有双侧腺母细胞瘤并伴有基于节段性肾小球硬化(暗示弗雷泽综合征)的延迟进行性肾衰竭的46,XY DSD患者被鉴定为新的SRY错义突变和WT1基因KTS剪切位点突变[21]。
3展望
随着性腺发育相关新基因和新机制的发现,出现了一些新的dsd概念。[22]发现了46,XX型糖尿病和46,XY型糖尿病患者的基因突变。例如,由于基因剂量效应、基因调控区的缺失或插入以及非性腺基因的异位表达,已经发现导致DSD的基因突变。随着DSD遗传原因的进一步确定,这些疾病的诊断将变得更加准确。
参考
[1]小野男,哈雷VR。性别发展障碍:新基因,新概念[。Nat Rev内分泌学,2013,9 (2) :79-91。
[2]北村光,柳泽男,杉山男,等. ARXcauses突变导致小鼠前脑和睾丸的异常发育和人类生殖器异常的X连锁无脑畸形[.Nat Genet,2002,32 (3) :359-69。
[3]吉本斯RJ1,皮克茨DJ,维拉德L,等。一个假定的全球转录调控因子的突变导致与α地中海贫血(ATR-X综合征)相关的精神发育迟滞[。牢房,1995年,24;80 (6) :837-45。
[4]离子阿,特尔维,肖申JL,等。一个新的突变在假定的脱氧核糖核酸解旋酶Xh2负责男性到女性的性别逆转与非典型形式的ATR-X综合征[。《遗传》,1996,58 (6) :1185-91。
[5]巴格里-法姆,阿根塔罗,斯温根,等.在ATR-X综合征小鼠模型中增殖的支持细胞和雄激素受体功能的存活缺陷[.哼摩尔热奈特,2011,20 (11) :2213-24。
[6]克拉克·阿姆,加兰·KK,拉塞尔·罗德。沙漠刺猬(Dhh)基因是小鼠睾丸中成年型睾丸间质细胞形成和肾小管周围细胞及生精小管正常发育所必需的。[。Biol Reprod,2000,63 (6) :1825-38。
[7]康托·普,维尔奇斯·福,斯?沙漠刺猬基因在性腺混合发育不良患者中的杂合突变[。《摩尔哼声再现》,2005,11 (11) :833-6。
[8][8]李迪格,郝特,温士奇,等. DMRT1causes46的部分缺失,XY睾丸性发育障碍[.《欧洲内分泌学杂志》,2012,167 (1) :119-24。
[9]洛伦?引用该论文李建华,李建民,等。gata4中功能缺失突变导致人类睾丸发育异常[。美国国家科学院学报,2011,108 (4) :1597-602。
[10]特沃西安SG,阿尔布雷特KH,克里斯平JD,等.小鼠性腺分化,性别决定和正常Sry表达需要转录伙伴GATA4和FOG2之间的直接相互作用]。发展,2002,129 (19) :4627-34。
[11]沃伦,博加尼,西格普,等。缺乏编码MAPK信号成分基因的小鼠睾丸发育的微小异常,MAP3K1。PlO S1,2011,6(5):19572。
[12]艾奇曼JC,伊藤,伊藤,伊藤,等。编码类固醇生成因子-1的基因突变导致人类XY性逆转和肾上腺衰竭[。Nat Genet,1999,22 (2) :125-6。
[13]怀特,休伊特,图尔比特,等.与性发育的46,XY障碍相关的野生型体内多外显子缺失[.《欧洲遗传学杂志》,2012,20 (3) :348-51。
[14]品牌?马默1(包含1个主谋样结构域)纯合功能增益错义突变,在一个男性女性中引起46,XX性发育障碍,[,2011.707,129-131。
[15]叶安生,池田勇,贾敏等.类固醇生成因子1的细胞特异性敲除揭示了其在性腺功能中的重要作用[.摩尔内分泌学,2004,18 (7) :1610-9。
[16]丰塞卡DJ,奥赫达D,拉哈尔B,等。2突变可能导致特发性卵巢早衰[。Transl Res,2012,160 (5) :384-8。
[17]帕尔玛,拉迪奥,维达尔五世,等。脊椎神经在性别决定,皮肤分化和恶性肿瘤中是必不可少的[。Nat Genet,2006,38 (11) :1304-9。
[18]查索,兰克,格雷瓜尔,等。通过Rspo1激活β-连环蛋白信号控制哺乳动物卵巢的分化[。哼摩尔热奈特,2008,17 (9) :1264-77。
[19]托马塞里,梅吉诺尼,林磊,等.人RSPO1/反应蛋白1在卵巢早期发育过程中表达,并增强β-连环蛋白信号[.PlO S1,2011,6 (1) :e16366。
[20]萨顿·E,休斯·J,怀特·S,等.在小鼠和人类中鉴定SOX3as为XX雄性逆转基因[·J]。《临床投资》,2011,121 (1) :328-41。
[21]赫姆斯,范德兹万YG,普鲁德,等.一名46,XY女性双侧性腺母细胞瘤患者,一种新的SRYmissense突变与WT1KTS剪接位点突变相结合,[.PlO S1,2012,7 (7) :e40858。
[22]巴桑布·阿,莱迪·斯,韦阿克·普,等.鉴定性发育障碍新遗传标记的新技术(DSD)[·杰]。性发展,2010,4 (4-5) :213-24。


